




Abstract
Inflammatory myofibroblastic tumor is a rare tumor deriving from mesenchymal tissue. Approximately 50% of inflammatory myofibroblastic tumors harbor an anaplastic lymphoma kinase fusion gene. Pulmonary inflammatory myofibroblastic tumors harboring tropomyosin3-anaplastic lymphoma kinase or protein tyrosine phosphatase receptor-type F polypeptide-interacting protein-binding protein 1-anaplastic lymphoma kinase have been reported previously. However, it has not been reported that inflammatory myofibroblastic tumors harbor echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase fusion gene which is considered to be very specific to lung cancers. A few tumors harboring echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase fusion gene other than lung cancers have been reported and the tumors were all carcinomas. A 67-year-old man had been followed up for a benign tumor for approximately 3 years before the tumor demonstrated malignant transformation. Lobectomy and autopsy revealed that an inflammatory myofibroblastic tumor harboring echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase fusion gene had transformed into an undifferentiated sarcoma. This case suggests that echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase fusion is an oncogenic event in not only carcinomas but also sarcomas originating from stromal cells.
INTRODUCTION
Inflammatory myofibroblastic tumors (IMTs) categorized as a subgroup of inflammatory pseudotumors (IPTs) are rare tumors that originate from mesenchymal tissue. IMTs are considered to be benign or low-grade malignant tumors, and a subset of these tumors exhibit recurrence, distant metastasis or malignant transformation (1–3). IPTs have various histological features; therefore, distinction of IMT from other mesenchymal tumors is difficult in a partial biopsy. However, anaplastic lymphoma kinase (ALK) fusion gene abnormalities in IMTs are a specific finding that differentiates these tumors from other tumors. Pulmonary IMTs harboring tropomyosin3-anaplastic lymphoma kinase (TPM3-ALK) or protein tyrosine phosphatase receptor-type F polypeptide-interacting protein-binding protein 1-anaplastic lymphoma kinase (PPFIBPI-ALK) have been reported previously (4,5), while echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) fusion has been considered largely specific to lung adenocarcinoma and an IMT harboring EML4-ALK fusion gene has not been reported.
CASE REPORT
A 67-year-old man who had cough from December 2006 visited a clinic and was referred to our hospital for a solid tumor that was revealed in a chest X-ray. A chest computed tomography (CT) scan showed a solid tumor in the right upper lobe of the lung with clear margins and a maximum diameter of 4.3 cm (Fig. 1A). Laboratory tests, including serum tumor markers, showed no abnormalities. Cytology at bronchoscopy in February 2007 gave negative results. He was followed up for a benign tumor that shrank slightly to 4.0 cm (Fig. 1B) until September 2009, while 18F-fluorodeoxyglucose (FDG)-positron emission tomography (FDG-PET) showed FDG uptake in the tumor (Fig. 1E). In October 2009, pathological findings on bronchoscopy showed the tumor contained spindle-shaped myofibroblasts and fibroblasts and infiltration of various inflammatory cells. Spindle cells had no cellular atypia or mitotic figures (Fig. 2A). There were no ALK-positive cells on immunostaining (Fig. 2B), although a few split signals of ALK gene on spindle cells were detected by the fluorescence in situ hybridization (FISH) study (Fig. 2C).
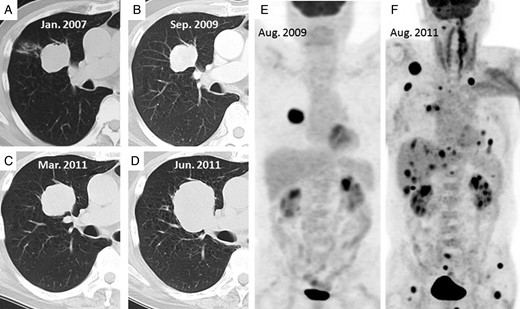
(A–D) Computed tomography (CT) examinations of the tumor in January 2007, September 2009, March 2011 and June 2011, respectively. (E) In August 2009, abnormal 18F-fluorodeoxyglucose (FDG) uptake is shown only in the tumor. (F) A FDG-positron emission tomography (PET) examination shows multiple metastases in August 2011 after recurrence.
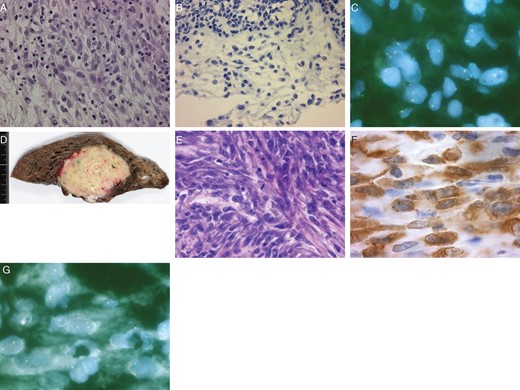
(A–C) Specimens on bronchoscopy in October 2009. (A) A picture of hematoxylin and eosin (H–E) stain shows that spindle cells have no cellular atypia or mitotic figures (×400). (B) ALK-positive spindle cells are not detected immunochemically (×400). (C) A fluorescence in situ hybridization (FISH) study shows that split signals of the ALK gene are already detected in a few spindle cells which are benign pathologically in October 2009. The red signal represents the 5′ end and the green signal represents the 3′ end of the ALK gene. (D–G) Lobectomy specimens in July 2011. (D) A macroscopic image of the lobectomy specimen shows that the tumor is whitish-yellow. (E) A picture of H–E stain shows that many spindle cells have large atypical nuclei with distinct nucleoli and mitosis is also detected (×400). (F) ALK is expressed in the cytoplasm of the spindle cells (×1000). (G) Split signals of ALK gene are detected easily by the FISH study in July 2011, compared with that in October 2009.
The patient underwent CT-guided biopsy in November 2010, because the tumor had increased to 4.3 cm. Pathological findings were almost identical to those of the previous examination, i.e. presence of spindle cells, infiltration of inflammatory cells and absence of epithelial cells on immunostaining. Prednisolone (40 mg/day) was administered, and the tumor responded very weakly, decreasing from 4.8 to 4.5 cm in diameter (Fig. 1C).
However, it increased rapidly to 6.1 cm (Fig. 1D) after the patient stopped steroid therapy voluntarily. He underwent right upper and partial middle lobectomy and mediastinal lymph node dissection in July 2011. A lobectomy specimen showed that the tumor was whitish-yellow with a maximum diameter of 6.3 cm (Fig. 2D) and had metastasized to the mediastinal lymph nodes. Pathological examination showed that the tumor contained spindle cells and polymorphic cells, with infiltration of inflammatory cells, invading the pulmonary vein. Many spindle cells had large atypical nuclei with distinct nucleoli and mitosis was 5–10 per 10 high-power fields (HPFs) (Fig. 2E). Immunohistochemically, the cells were negative for epithelial markers, such as CAM5.2, CK5/6, CK7, CK14, EMA, AE1/AE3 or TTF-1. However, ALK (Fig. 2F) and vimentin were expressed in the cytoplasm of the spindle cells, a subset of which was positive for p53. FISH indicated that the spindle cells had split signals of ALK gene (Fig. 2G). The tumor was diagnosed as IMT that had transformed into an undifferentiated sarcoma.
The patient suddenly experienced disorientation, right hemiplegia and restlessness approximately 2 months after lobectomy. MRI, FDG-PET and CT examinations revealed local recurrence, and brain, multiple pulmonary, subcutaneous and liver metastases (Fig. 1F). Whole-brain radiation therapy had no effect on brain metastases, and he died approximately 1 month after recurrence. Autopsy was performed. Autopsy revealed multiple organ metastases. Histopathologically, the tumor consisted of proliferation of sarcomatoid cells and had no evidence of epithelial components on multiple samples. Direct PCR sequencing revealed that the tumor harbored EML4-ALK fusion gene (Fig. 3).
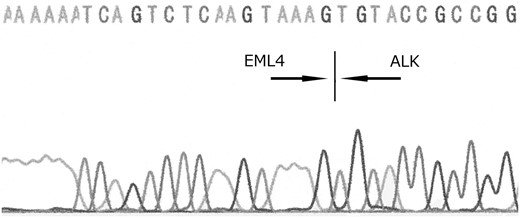
EML4-ALK fusion gene (variant 5a) is detected in the tumor tissue by direct sequencing.
DISCUSSION
IMTs are rare tumors originating from mesenchymal tissue and involve various morphologies, such as a combination of proliferation of spindle cells and infiltration of inflammatory cells. Recently, IMT is categorized as a subgroup of IPT, which is an independent disease because of the presence of an ALK gene abnormality. Approximately 50% of IMTs harbor an ALK fusion gene, which is normally found in anaplastic large-cell lymphoma, non-small-cell lung cancer (NSCLC) and large B-cell lymphoma, as well as IMT (6). Pulmonary IMTs harboring TPM3-ALK or PPFIBP1-ALK have been reported previously (4,5). EML4-ALK is known to be an abnormality of NSCLC (7), with a prevalence of 1–5% (8–10). Soda et al. (7) reported the oncogenicity of EML4-ALK fusion gene abnormalities in transgenic mice with pulmonary epithelial cells expressing the fusion gene, and revealed that the mice developed lung adenocarcinoma a few weeks after birth. No study has shown EML4-ALK expression in pulmonary stromal cells. This case implies that the etiology of IMT may be an ALK fusion gene in lung stromal cells regardless of the fusion partner. However, few reports have revealed the genetic origin of the fusion partner of ALK in pulmonary IMTs. No expression of the EML4-ALK fusion gene was observed in tumors other than NSCLC in some studies (10,11). In contrast, the EML4-ALK fusion gene has been reported in colorectal and breast cancers (12) and a renal cancer (13). In general, however, it is believed that this gene abnormality is largely specific for lung cancer.
In terms of the correlation between genotype and phenotype of EML4-ALK, it might be speculated that this case was a sarcomatoid carcinoma without epithelial component mimicking IMT. Sarcomatoid carcinomas with EML4-ALK fusion have been previously reported (14). We have diagnosed this case as IMT by typical pathological features with immunohistochemical findings. The tumor was already larger than 4 cm at the first visit and did not grow for approximately three years. Pathological findings of biopsies, surgical specimen and autopsy materials revealed no evidence of an epithelial component. Retrospectively, we performed the FISH study on biopsy tissue collected in October 2009 and found out the abnormal ALK gene signal in spindle cells without cytological atypia or epithelial markers by immunostaining. On the basis of these findings, we concluded that the tumor in this case was not a sarcomatoid carcinoma, but an IMT harboring EML4-ALK fusion gene and subsequently demonstrated malignant transformation into a sarcoma. The malignant features such as recurrence, metastasis or malignant transformation are considered to be rare events in IMTs. Some studies have described sarcomatous transformation (2) or implied that ALK had a positive effect on the outcome in IMT because IMTs expressing ALK did not develop distant metastases (1,15). This case suggests that EML4-ALK may affect tumorigenesis of IMT, and additional other gene abnormalities such as p53 may introduce malignant transformation because p53 expression is rare in IMT but may be useful in identifying IMT developed malignant transformation or recurrence (3).
It has been often considered that EML4-ALK fusion is specific to lung adenocarcinoma because of the correlation between the genotype and the phenotype. However, this case suggests that EML4-ALK fusion gene also affects stromal cells and leads to IMT development.
Conflict of interest statement
None declared.

{kind=link}
{kind=link}
{kind=link}